Biliary Atresia: Symptoms, Causes, Types, Risk Factors & Complications
PACE Hospitals
Overview | Prevalence | Types | Symptoms | Risk Factors | Causes | Complications | Diagnosis | Treatment | FAQs | When to Consult a Doctor
What is Biliary Atresia?
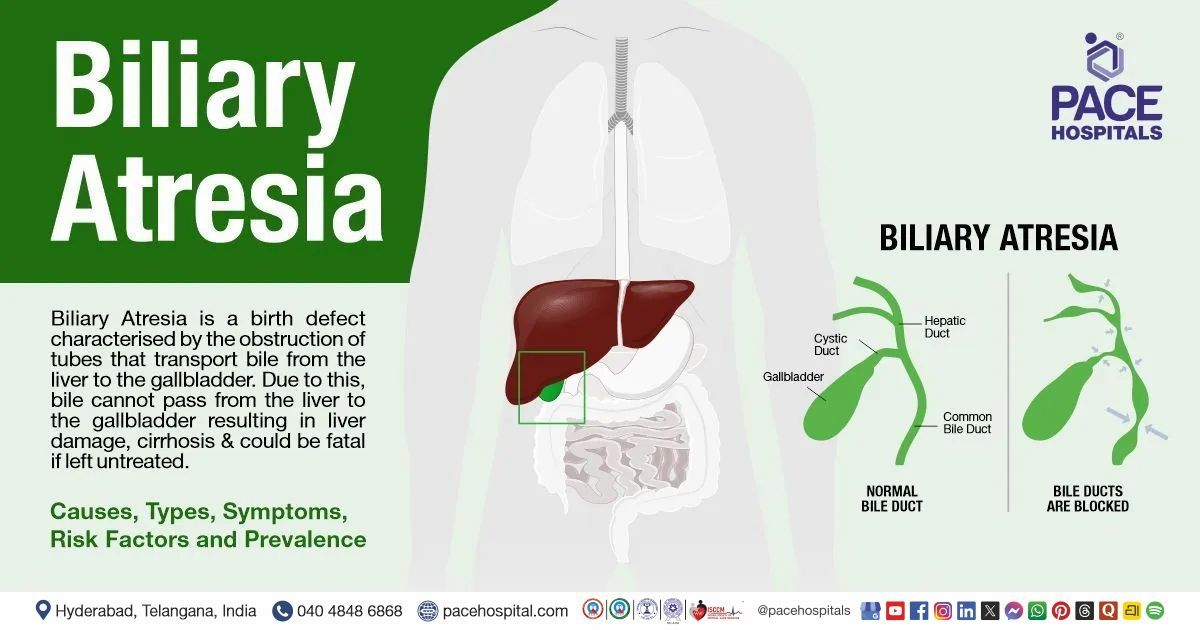
Biliary atresia is a congenital disorder of obstruction in the tubes (ducts) that transport bile from the liver to the gallbladder due to abnormal development of the bile ducts within or outside the liver. In infants with biliary atresia, bile cannot pass from the liver to the gallbladder, resulting in liver damage and cirrhosis and could be fatal if left untreated.
It is also known as extrahepatic ductopenia or progressive obliterative cholangiopathy.
Biliary Atresia Prevalence
The most common of the rare neonatal cholestatic syndromes, biliary atresia occurrence is seen in about one out of every 8,000-12,000 live births globally. It is mainly seen in female infants.
Biliary Atresia Types
There are various types of biliary atresia classification.
Depending on its occurrence, there are two types of biliary atresia, which are:
- Perinatal biliary atresia: The most common type of biliary atresia appears 2-4 weeks after the baby is born.
- Foetal biliary atresia is less commonly seen and appears in the baby while developing in the mother’s womb (gestational period).
Based on the location of the block and degree of pathology, the Kasai system of biliary atresia classification divides the ailment into three main types based on the anatomical position of the block (obliteration):
- Type I: obliteration is seen only in the common bile duct, while the proximal bile ducts are intact
- Type II: Type 2 has IIa and IIb. Type IIa describes the obliteration of the common hepatic duct with the intact common bile duct.
- Type IIb: The cystic bile duct, the common bile ducts and the common hepatic duct are obliterated.
- Type III is the most common form, accounting for more than 90% of cases. Here the cystic bile duct, the common bile ducts, the hepatic ducts, and the intrahepatic ducts are obliterated.
Type III is also called extrahepatic biliary atresia. Extrahepatic biliary atresia is most commonly associated with total and permanent cholestasis. Here the bile duct could be partially or blocked between porta hepatis and the duodenum, and the worldwide incidence could range from 1:8,000-1:10,000.

Biliary Atresia Symptoms
At birth, infants with biliary atresia may look healthy, but jaundice develops during the second or third week of life. The newborn may gain weight generally during the first month but will then lose weight, develop irritability, and have severe neonatal jaundice. The parents and caretakers are responsible for considering and comprehending the child's anomalies.
Other symptoms of biliary atresia may include:
- Splenomegaly (enlarged spleen) - The complications of splenomegaly include anaemia (reduction of red blood cells) and leucopoenia (a reduction in the count of white cells), leading to more frequent infections.
- Porphyria (dark urine) - The complications of may include chronic kidney and liver failure, or liver cancer.
- Steatorrhea (too much fat in stools causing - floating stools) –bile produced in the liver emulsifies oils and fats, thus making them digestible with help of lipase. Since bile is not produced, the fats are excreted undigested.
- Foul-smelling stools (too much fat in stools) – Due to excretion of undigested fats
- Acholic stools (pale or clay-coloured stools)
- Slow growth
- Slow or no weight gain
The paediatricians particularly look for “biliary atresia poop", one of the confirming factors of steatorrhea.

Biliary Atresia Risk factors
The reason for biliary atresia occurrence is unknown, but it can be caused by immune, infectious/toxic, and genetic factors.
More of correlations rather than risk factors, a 2007 study published in the American Journal of Medical Genetics demonstrated the assessment from the statistics of the National Birth Defects Prevention Study (NBDPS) of America to understand the potential correlations which could have caused this ailment. It was shown that biliary atresia is more likely to occur.
- In infants from non-Hispanic Black mothers.
- In preterm infants.
- In preterm infants from older women.
- Among infants born in autumn than in summer.
- In infants whose mothers had a respiratory illness but it must be understood that the germs involved in respiratory disease are not those most likely to cause biliary atresia.
- In infants of mothers who suffered malnutrition of vitamin E, copper, iron, phosphorus, and beta tocopherol.
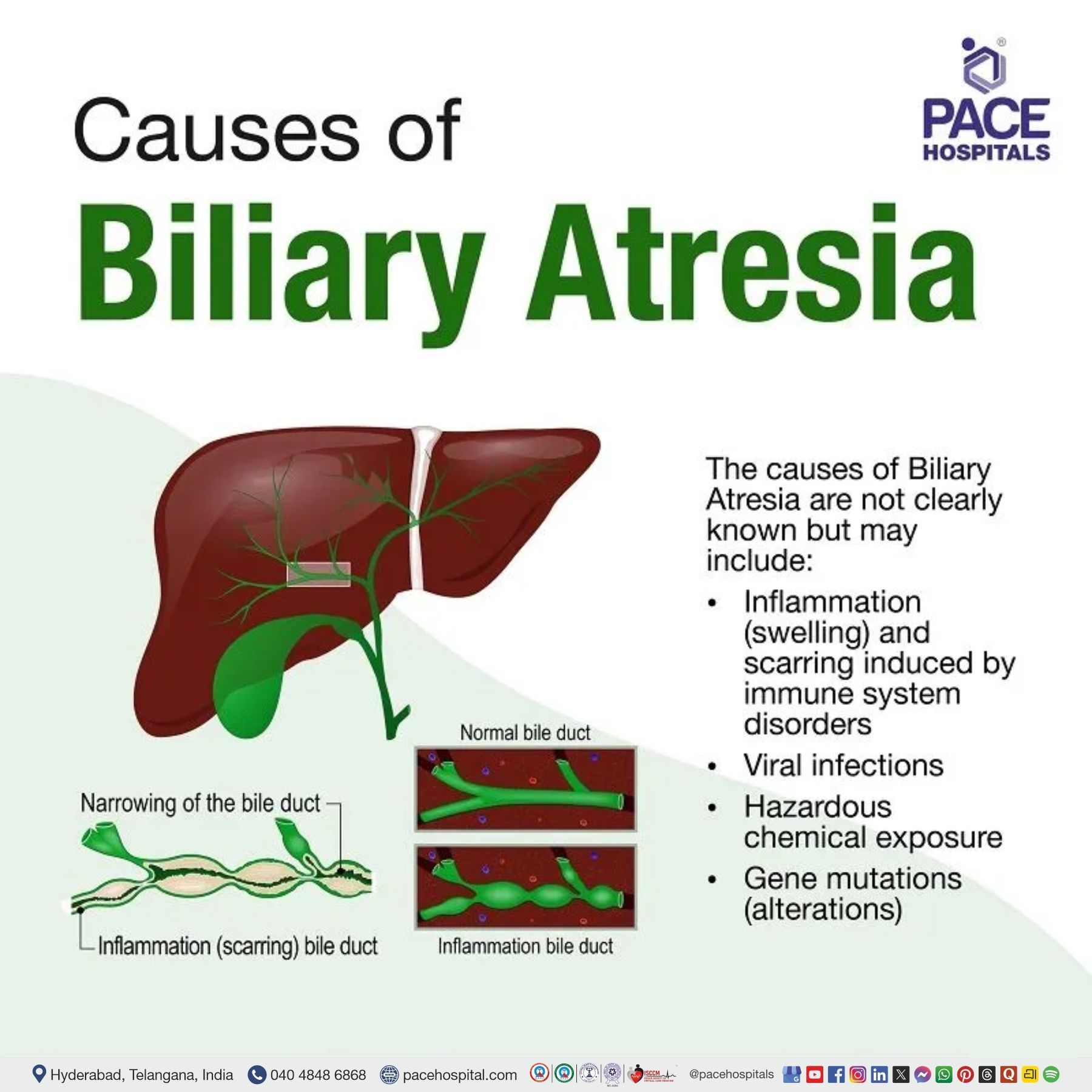
Biliary Atresia Causes
The causes of biliary atresia are not known, but may include the following:
- Inflammation (swelling) and scarring induced by immune system disorders
- Viral infections
- Hazardous chemical exposure
- Gene mutations (alterations). (Biliary atresia is not passed on from parents to children.)
Although biliary atresia is not an inherited disease, a minority of cases could be caused due to morphogenetic defects (faultlines developed during the growth of the biliary tree & liver during pregnancy). A prenatal ultrasound exposing a cyst in the biliary system could diagnose the ailment during gestation.
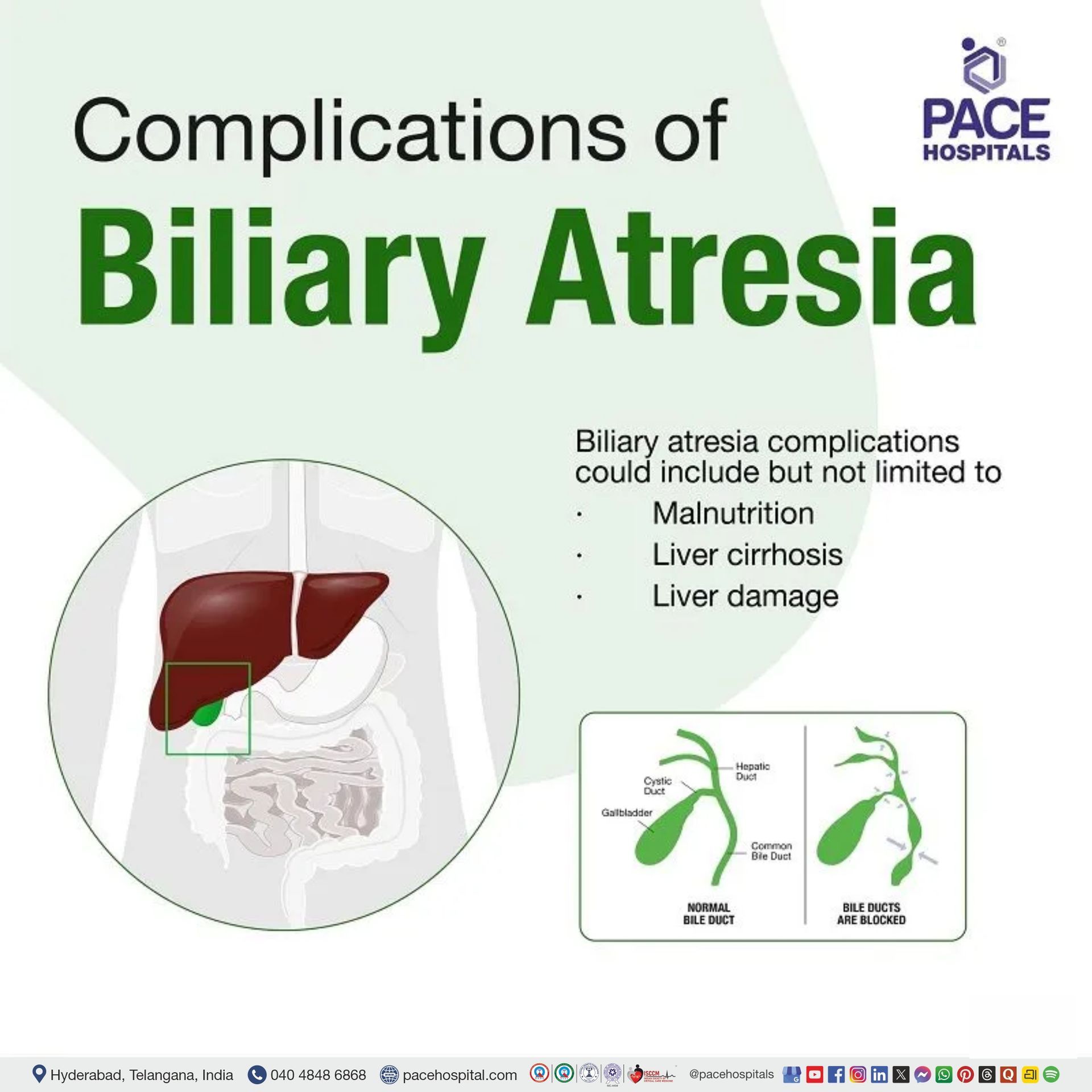
Biliary Atresia Complications
Children suffering from biliary atresia consequently suffer from a reduction of bile flow into the small intestine leading to malnutrition, culminating with liver damage. The paediatricians may develop a unique eating plan containing enough nutrients, calories and supplements after consulting dietitians.
Biliary atresia splenic malformation syndrome (BASM)
About 10% of infants suffering from biliary atresia also display other innate life-limiting fibro-obliterative anomalies such as:
- Biliary atresia splenic malformation (BASM) syndrome
- Over 80% of the infants suffering from biliary atresia splenic malformation demonstrate various other splenic abnormalities such as:
- Polysplenia (2 or more spleens with numerous congenital abnormalities in the abdomen and chest),
- Double spleens (presence of 2 spleens)
- Asplenia (no spleen) with or without
- Situs inversus (organs of the and abdomen and chest are formed in a mirror image of normal human anatomy)
- Preduodenal portal vein (the portal vein passes anterior to the duodenum instead of posterior to the duodenum)
- Absent inferior vena cava (large vein carrying deoxygenated blood into the heart from the lower body)
- Malrotation (a congenital disability where the intestines do not completely rotate into their normal final position during development) and
- cardiac anomalies (heart defects)
Biliary atresia diagnosis
The biliary atresia investigations include the following:
- History and Physical examination
- Liver function tests
- Ultrasonography (biliary atresia ultrasound)
- Cholangiography (X-ray examination of the bile ducts, used to locate and identify an obstruction)
- Liver biopsy (biliary atresia gold standard diagnosis)
- Hepatobiliary scintigraphy
Why Choose PACE Hospitals?
Expert Super Specialist Doctors
Advanced Diagnostics & Treatment
Affordable & Transparent Care
24x7 Emergency & ICU Support
Biliary atresia treatment
Biliary atresia (BA) is an obstructive neonatal cholangiopathy which leads to the development of end stage liver disease within the first 6–12 months.
Presently, there is no medical therapy to cease the continuity or reverse this cholangiopathy.
The hepatoportoenterostomy, introduced by Kasai in 50’s decade, till date remains the only glaring potential corrective operative procedure. The Kasai procedure results reinvigorated the biliary flow from the liver to the intestine. Untreated biliary atresia could lead to hepatic failure, progressive hyperbilirubinemia, cirrhosis, and death.
Frequently asked questions
When to Consult a Doctor for Biliary Atresia?
Consult a doctor immediately if a baby has persistent jaundice beyond two weeks of age, pale or clay-coloured stools, dark urine, or poor weight gain, as these may be signs of Biliary Atresia, a serious liver condition requiring early treatment.
Warning Signs:
- Jaundice lasting longer than 2 weeks after birth
- Pale, grey, or clay-coloured stools
- Dark urine
- Poor weight gain or growth
- Swollen abdomen
- Enlarged liver
- Easy bruising or bleeding
- Persistent irritability
See a paediatrician, a pediatric hepatologist, a pediatric surgeon, or a pediatric gastroenterologist if these symptoms appear. To confirm the diagnosis and select the best course of treatment, the physician may recommend blood tests, liver function tests, abdominal ultrasound, hepatobiliary imaging, and liver biopsy.
Share on
Request an appointment
Fill in the appointment form or call us instantly to book a confirmed appointment with our super specialist at 04048486868
Appointment request - health articles
Recent Articles